晶体与晶胞
预计阅读时间: 35 分钟化学键与晶体
化学键
分子内部相邻原子间强烈的相互作用,称为化学键,主要决定了物质的化学性质。化学反应的过程实质是,旧化学键的断裂(吸收热量),和新化学键的生成(放出热量)。

化学键分为离子键、共价键、金属键,其中有配位键作为一个特殊的共价键。
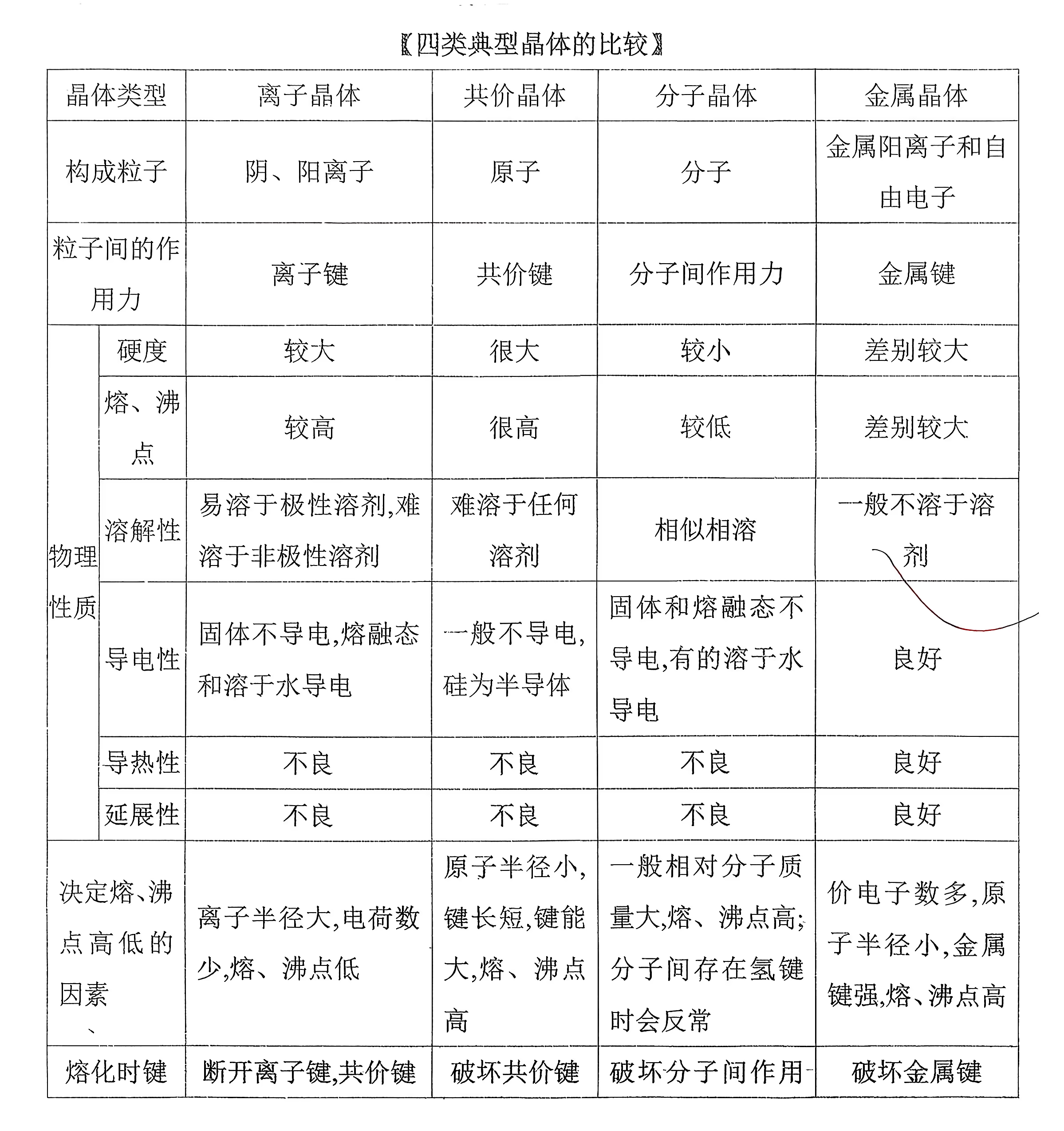
晶体与非晶体的比较:
晶体与非晶体的区分,间接方法是看是否有固定的熔点,但是这不是最科学的方法,严谨来讲需要对固体进行 X 射线衍射实验。得到晶体的途径有,熔融态物质凝固,气态物质凝华,溶质从溶液中过饱和析出。
测定晶体结构最常见的仪器是 X 射线衍射仪。在晶体的 X 射线衍射实验中,当单一波长的 X 射线通过晶体时,X 射线与晶体中的电子相互作用,会在记录仪上产生分离的斑点或明锐的衍射峰。有衍射图形获得的晶体结构的信息包括晶胞的形状和大小、分子或原子在微观空间有序排列呈现的对称类型、原子在晶胞中的数目和位置等。
离子晶体
离子键:阴阳离子之间强烈的静电作用(吸引和排斥),称为离子键,活泼金属(或铵根)与非金属之间易形成离子键。
离子化合物:含有离子键的化合物称为离子化合物,绝大多数盐( 为特例)、强碱、活泼金属氧化物、硫化物为离子化合物。
粒子晶体:离子再空间上周期性重复排列。
离子键性质:
-
无饱和性:在离子化合物中,每个离子周围最邻近的带异性电荷离子数目的多少,取决于阴、阳离子的相对大小。
只要空间条件允许,阳离子将吸引尽可能多的阴离子排列在其周围,阴离子也将吸引尽可能多的阳离子排列在其周围,以达到降低体系能量的目的。
-
无方向性:离子键的实质是静电作用,离子的电荷分布通常被看成是球形对称的,因此一种离子可以对不同方向的带异性电荷的离子产生吸引作用。
离子键的极化:
-
基本上呈球对称的离子本身所带有的电荷会形成一个电场,阴、阳离子在相互的电场作用下,会使离子内的电子分布发生相对偏移,这种在电场的作用下产生的离子中的电子分布发生偏移的现象称为离子极化。
-
离子极化可能导致阴、阳离子的外层轨道发生重叠,从而使得许多离子键不同程度地带有一些共价性,继而导致键长缩短、键能增加,甚至出现结构变异。
-
例如:从 到 ,离子键愈来愈倾向于共价形式, 的键长几乎等于半径之和,但是半径之和增大,键长的增大却跟不上半径之和,到 中半径之和已经明显大于键长,导致其成为以共价键为主的结构,卤化银溶解度自上到下减小。
离子键强度:
-
离子键强度越大,键能越大,熔沸点越高。
-
离子键的破坏:发生化学变化、电离(溶于水或熔融)。
-
晶格能是指 离子晶体气态完全解离为正负离子所吸收的能量,或反之释放的能量。
-
玻恩-哈伯循环是一种利用盖斯定律间接计算离子化合物晶格能的方法。由于晶格能不能直接测量,玻恩-哈伯循环通过分解反应路径,将复杂的能量变化拆解为多个可实验测量的步骤,最终通过能量守恒计算晶格能。
玻恩-哈伯循环将一个简单的化学反应拆分为若干步,根据盖斯定律他们的焓变相等,而整个形成的循环中只有晶格能未知,可以达到测量晶格能的效果,整个过程如下所示:
根据盖斯定律,上述所有焓变加起来就是 的标准摩尔生成焓 ,列出式子:
拓展到多价离子:
-
电离能拓展为第一 、第二 两级电离。
-
电子亲和能拓展为第一 、第二 两级亲和。
-
注意电子亲和能有可能为正值,取决于产物稳定性。
晶格能的影响因素:
- 离子电荷:电荷越大,晶格能越大(如 )。
- 离子半径:半径越小,晶格能越大(如 )。
- 离子排列方式:紧密堆积结构,晶格能更高。
金属晶体
定义:
-
金属原子的电离能低,容易失去电子而形成阳离子和自由电子,阳离子整体共同吸引自由电子而结合在一起。
-
这种金属阳离子和自由电子之间强烈的相互作用就是金属键,是一种遍布整个晶体的离域化学键。
-
金属键可看成是由许多原子共用许多电子的一种特殊形式的共价键,但与共价键有着明显不同。
-
金属键既没有方向性也没有饱和性,成键电子可以在金属中自由运动,使得金属呈现出特有的属性。
成键微粒:
-
金属阳离子和自由电子(金属晶体中,金属失去电子形成金属阳离子和电子,没有阴离子)。
-
通过金属键延伸形成的晶体为金属晶体,其中不存在单个分子,金属阳离子被自由电子所包围。
金属键的强弱:
-
金属阳离子半径:半径越小,键越强。
-
离子带电荷数(单位体积内的自由电子数):带电荷数越多,键越强。
-
金属阳离子半径越小、所带电荷越多、自由电子越多、金属键越强、熔点越高,硬度越大。
金属键的递变规律:
-
同主族元素:从上到下原子半径依次增大,金属键依次减弱。
-
同周期元素:从左到右价电子数增大,原子半径依次减小,金属键依次增强。
金属键对金属的影响:
-
金属不透明,具有金属光泽:当可见光照射到金属表面上时,固态金属中的"自由电子"能够吸收所有频率的光并很快放出,使得金属不透明并具有金属光泽。
-
金属具有良好的导电性:金属内部自由电子的运动不具有方向性,在外加电场的作用下,金属晶体中的"自由电子"沿着导线由电源的负极向电源正极发生定向移动而形成电流。温度越高,金属的导电性能越差。
-
金属具有良好的导热性:当金属中有温度差时,通过不停运动着的"自由电子"通过自身与金属阳离子间的碰撞,把能量由高温处传向低温处。
-
金属具有良好的延展性:当金属受到外力作用时,晶体中的各原子层就会发生相对滑动,但排列方式不变,金属晶体中的化学键没有被破坏。
金属的熔沸点:
-
金属熔沸点整体来说没有一般规律。
-
最低的是铯单质,最高的是钨单质。
-
碱金属金属从上到下熔沸点递减。
共价晶体
相邻原子间以共价键结合而形成的具有空间立体网状结构的晶体称为共价晶体。共价晶体总是具有空间网状结构,依靠共价键在空间中、三维上无限延伸,可以看为一个巨型的分子,因此链状不能算共价晶体,那通常是聚合物晶体(或称为结晶性聚合物),共价晶体没有分子式,其化学式一般指的是最简式。
共价晶体的硬度很大,同时熔沸点很高,共价晶体的熔点一般指的是将共价键破坏,而沸点通常是原子化了,高中不太考虑。对于结构相似的共价晶体来说,原子半径越小、键长越短、键能越大,晶体的熔点就越高。
计算共价键数量:
-
无机物:相同看一半,不同都要看(详见晶胞部分)
-
有机物:碳四、氢一、氧二,成键加和减半,例如 有 ,对于 有 。
共价晶体更多详情见共价键模型,这里给出一部分。
-
半径越小、键长越短,键能越大,共价键越强,熔沸点越高,硬度通常越大。
-
例如 ,就是由分子半径决定的。
典型共价晶体结构模型:通常情况下,晶体硅、晶体锗、晶体硼、碳化硅、二氧化硅、氮化硼等都属于共价晶体。
-
金刚石的结构:
-
在晶体中,每个碳以 个单键对称的与相邻的 各碳原子相结合,形成正四面体结构,这些正四面体向空间发展,构成彼此联结的立体网状结构。
-
晶体中,相邻碳碳夹角为 ,碳原子采用 sp^3^ 杂化。
-
最小环上有 个碳原子,晶体中碳原子个数与碳碳键数之比为 。
-
晶体中碳碳键很短,键能很大,故金刚石的硬度很大,沸点很高。
-
-
二氧化硅的晶体结构:
-
在晶体中,每个硅原子周围结合 个氧原子,同时每个氧原子与 个硅原子结合。
-
在晶体中,位于四面体中心的是硅原子,位于四面体顶点上的是氧原子, 二氧化硅中有 硅氧键。
-
二氧化硅晶体中最小环上的原子数是 ,包括 个氧原子和 各硅原子。
-
-
碳化硅的晶体结构:碳化硅的晶体结构类似于金刚石的晶体结构,其中碳原子和硅原子的位置是交替的,所以在整个晶体中,硅原子与碳原子个数之比为 。
注意一下几点:
-
共价晶体的构成微粒是原子,只存在共价键,不存在其他作用力。
-
共价晶体的化学式表示其比例组成,晶体中不存在分子。
-
共价晶体一般都熔点高、硬度大、不溶于溶剂,一般不导电。
分子晶体
分子间通过分子间作用力结合形成的晶体,在空间中通过氢键、范德华力等分子间作用力延伸。部分非金属单质、金属氧化物、金属氢化物、几乎所有的酸、大部分有机物,都是分子晶体。
-
碘晶体:一个长方形,顶点和面心上各有一个 分子,每个晶胞中有 个 分子,分子间以范德华力结合。
-
干冰晶体:面心立方,配位数 ,融化时化学键不断裂。
-
冰晶体:水分子之间的主要作用力是氢键,当然也存在范德华力。氢键有方向性,它的存在迫使在四面体中心的每个水分子与四面体顶角的 各相邻水分子相互吸引。
分子晶体由于以比较弱的分子间作用力相互结合,因此一般熔点较低,硬度较小,在常温常压下经常是气体。一般来说,有氢键的分子晶体熔沸点更高,但是不排除范德华力反客为主成为主导因素,尤其是分子质量相差非常大的情况。在没有氢键且结构相似的情况下,一般分子质量越大,范德华力越大,分子晶体的熔沸点越高。
混合晶体
石墨晶体是层状结构,在每一层内,每个碳原子与临近的三个碳原子以共价键结合,形成无限的六边形网状结构。每个碳原子还有一个与碳环平面垂直的未参与杂化的 2p 轨道,并含有一个未成对电子,形成遍及整个平面的超大离域 键,电子可以在其中自由运动,因此石墨具有各向异性的导电性。同时,石墨的层与层之间以范德华力结合,因为范德华力远小于共价键,受力很容易散开,因此作为铅笔芯的材料。
物质组成的复杂性导致晶体中存在多种不同微粒以及不同的微粒相互作用。金属键、离子键、共价键、配位键等都是化学键的典型模型,但是,原子之间形成的化学键往往是介于经典模型之间的过度状态。
熔沸点比较:
-
一般来说,熔沸点规律为共价晶体 离子晶体 分子晶体(氢键 范德华力)。
-
同时,如果相对分子量足够大,范德华力也可以大于氢键的影响。
-
都是 XX 晶体, 半径更小, 键更短,共价键更强,熔沸点更高。
-
都是离子晶体,半径(谁小),电荷(谁大),(谁的)晶格能大,离子键更强,熔沸点更高。
注意:只写支持观点的原因。
晶体晶胞
晶胞是描述晶体结构的最小单元,晶体中晶胞的排列是无隙且并置的,也就是说,不能把一个旋转拼接的单元当作晶胞。

原子坐标:
-
以晶胞参数为单位长度。
-
原子坐标 满足 。
-
晶胞参数:晶胞通常来说是一个平行六面体,因此可以用 各参数来表示,包括三组棱长 和三组夹角 ,即晶格特征参数,简称晶胞参数。
均摊法数个数:
-
原子在晶胞中的占有率: 个晶胞完全包含该原子,则该原子对每个晶胞有 。
-
配位数:与一原子距离最近且相同的粒子个数。
满足配位数之比与原子个数之比成反比。
-
空间利用率
-
晶胞密度:
结构推断问题
微粒的电子总数
[TODO]
十电子微粒:
十八电子微粒:
常见配位数配合物
六配位:
核反应方程式
根据反应前后质子、中子(及电子)守恒,即质量数(左上角)、质子数(左下角)加和相等,判断元素序数。
人们常采用较短小精简的符号描述核反应,许多质量轻的粒子常缩写,例如 p 代表质子,n 代表中子,d 代表氘( 核,一个中子一个质子), 代表 粒子( 核,两个中子两个质子), 代表 粒子(电子), 代表 光子。
未成对电子个数
有 个未成对电子:
有 个未成对电子:
有 个未成对电子:
有 个未成对电子:
-
有 个未成对电子:。
-
有 个未成对电子:。
-
有 个未成对电子:(最多)。
有 个未成对电子:
-
区:IIA。
-
区:IIB。
-
区:〇族。
结构拓展
离域大 键
离域 键,或共轭大 键,简称大 键。
电子效应
电子效应(Electronic Effects)是连接结构化学(微观电子分布)与有机化学(宏观反应性能)的桥梁。简单来说,它描述了分子内电子云是如何分布的,以及这种分布如何决定分子的稳定性、酸碱性和反应活性。
电子效应本质上可以用分子轨道的离域来统一解释:
-
诱导效应 型分子轨道中,由于原子电负性不同导致的系数差异。
-
共轭效应 型分子轨道的离域程度。
-
超共轭效应 轨道与 轨道之间的轨道混合(orbital mixing)。
核心思想:所有电子效应本质上都是电子密度在分子中的重新分布,只是传递机制不同。理解这一点,就掌握了有机化学的"第一性原理"。
诱导效应
由于原子或基团电负性差异,引起 键电子云沿碳链发生偏移的现象。
吸电子诱导效应()与供电子诱导效应():
-
效应(吸电子,比 强地吸引 键电子):
规律:电负性越大、不饱和度越高、正电荷越多 越强
-
效应(供电子,比 弱地吸引 键电子):
经典应用一氯代乙酸的酸性比乙酸强:
的 效应 使羧基 键电子偏向分子内部 更易解离 酸性增强。
多氯取代后酸性更强( 叠加):
共轭效应
在共轭体系中, 轨道彼此平行重叠,电子离域(delocalization)形成离域分子轨道,电子云不再局限于两个原子之间。
从结构化学角度(Hückel 分子轨道理论):
对 1,3-丁二烯, 个碳的 轨道线性组合形成 个 分子轨道:
其中 为库仑积分, 为共振积分()。
(供电子共轭效应):基团向共轭体系提供电子密度。
含有孤对电子的基团与 体系共轭时通常表现为 。
注意:卤素虽然有 效应,但同时有 效应。
(吸电子共轭效应):基团从共轭体系吸取电子密度。
含有极化的多重键的基团(原子上有正电性)。
苯酚 vs. 环己醇:
原因:苯酚的 上的孤对电子与苯环发生 共轭,使 键电子偏移,且生成的酚氧负离子 的负电荷通过共轭离域到苯环上,大大稳定了共轭碱。
共振结构(负电荷分散到邻位和对位)
共振结构是描述共轭效应的另一种等效语言(Pauling):
以苯胺为例:
书写规则:
-
各共振结构中原子核位置不变,只改变电子分布。
-
共振结构必须是合理的 Lewis 结构。
-
各共振结构的未配对电子数相同。
-
主要共振结构满足:电荷分离最少、八隅体完整、负电荷在电负性大的原子上。
超共轭效应
键的 轨道与相邻的 轨道部分重叠,产生电子离域------ 超共轭。实质: 键电子与相邻 电子部分离域。
丙烯中:
个 的 轨道与 的 轨道发生超共轭 键略有伸长, 单键略有缩短。
超共轭效应的强度与 键数目成正比:
超共轭:正碳离子中,相邻 键与缺电子碳的空 轨道重叠。
这正是碳正离子稳定性顺序的根本原因之一。
丙烯与 反应:
中间体 碳正离子 有 个 超共轭,比 碳正离子 (仅 个 )更稳定。
场效应
取代基的偶极通过空间(不通过化学键链)对反应中心产生的静电影响。
邻位取代苯甲酸的酸性异常增强,不能仅用诱导效应解释,还涉及场效应------取代基偶极在空间上直接影响羧基。
顺-2-氯环己烷羧酸 vs. 反-2-氯环己烷羧酸的酸性差异:顺式更强( 通过空间更靠近 )。
诱导效应与共轭效应的竞争与协同:
-
方向一致的情况:
-
方向矛盾的情况:
核心原则:当两者矛盾时,需根据具体分子和反应情景分析。共轭效应一般强于诱导效应(但 等高电负性原子例外)。
电子效应的应用
芳香族亲电取代反应的定位规则:
卤素的特殊性:
-
效应使邻对位电子密度相对增加 邻对位定位。
-
效应使苯环整体电子密度降低 钝化苯环。
氯苯的亲电取代:速率 苯,但产物以邻对位为主
用共振结构理解:
负电荷只出现在邻位和对位 亲电试剂进攻这些位置。
碳正离子稳定性:
解释:烷基的 效应 和 超共轭效应 共同分散正电荷。
共轭碱 / 酸稳定性:
酸性比较范例:
反应活性与选择性:
Michael 受体的活性:
吸电子共轭效应越强 碳正电性越大 越容易受到亲核进攻。